Your First Workflow on Google Cloud Virtual Machine (Snakemake RNA-seq)
This Snakemake RNA-seq workflow maps read-pairs to a reference genome and quantify the produce to produce a transcript. Snakemake workflows are Python based, extended by declarative code which defines rules. The rules indicate how the output files are created from the input files.
Requirements:
Note
The requirements above are crucial to running this workflow. Please make sure you have them installed properly prior to running this workflow.
Download this tutorial:
$ sudo add-apt-repository universe
$ sudo apt update
$ sudo apt install subversion
#cloning this tutorial
$ svn checkout https://github.com/isb-cgc/RunningWorkflows-on-the-GoogleCloud/trunk/Snakemake-RNAseq
Starting folder:
Snakemake-RNAseq
|_________data
| |_________ggal_gut_1.fq
| |_________ggal_gut_2.fq
| |_________ggal.gtf
| |_________reference.fa
|_________step1
| |_________indexing.smk
| |_________Snakefile
|_________step2
| |_________therest.smk
| |_________Snakefile
|_________environment.yml
Setting up your Conda environment using the environment.yml file
The file environment.yml contains a list of all the softwares required to run this tutorial
$ cd Snakemake-RNAseq
$ conda env create --name tutorial --file environment.yml
$ conda activate tutorial
Note
Syntax: conda env create –name <yourEnvironmentName> –file environment.yml. For this tutorial <yourEnvironmentName> = tutorial
It should look like this:

Congratulations! You are now ready to run your first workflow using snakemake.
Step 1: Creating Index files from reference genome using hisat2-build
From folder rnaseq-snake:
$ cd step1
Let’s take a look at the 2 files in this folder: Snakefile and indexing.smk.
|_________step1
|_________indexing.smk
|_________Snakefile
Snakefile is the default name for snakemake’s script. The “snakemake” command will automatically find a file name Snakefile and execute it; indexing.smk is a helper file that will be called within the Snakefile.
Let’s run it by using:
$ snakemake
Snakemake will run and use hisat2 to build index files from file rnaseq-snake/data/reference.fa. All the index files will be moved to rnaseq-snake/step1/reference.
Let’s take a quick look at the code:
#Snakefile
rule targets:
input:
"index.done"
include: "indexing.smk"
The first rule’s input of a Snakefile (rule targets) will determine what’s the output of that workflow. In this case, index.done is the target file that snakemake will look for. Include: “indexing.smk” will call the following helper script:
Note
Only the very first Snakefile rule’s input behaves like this; other input will be the actual inputs of your workflow.
#Indexing.smk
rule indexing:
input:
"../data/reference.fa"
output:
touch('index.done')
shell:
"""
mkdir reference
hisat2-build {input} index
mv index.* reference/
"""
In Indexing.smk file we have an actual input “../data/reference.fa” and the output section tells snakemake to create an empty file “index.done”, which is the file that the first rule will check to make sure that this helper script actually run. Then the shell script is executed as follows: a folder called reference got created, then Hisat2 created index files from the fasta file, and then all the index files got moved to the reference folder.
After Step 1:
rnaseq-snakemake
|_________data
| |_________ggal_gut_1.fq
| |_________ggal_gut_2.fq
| |_________ggal.gtf
| |_________reference.fa
|_________step1
| |_________indexing.smk
| |_________Snakefile
| |_________[index.done]
| |_________[reference]
| |_________[index.1.ht2]
| | [(2-7)]
| |_________[index.8.ht2]
|_________step2
| |_________therest.smk
| |_________Snakefile
|_________environment.yml
Step 2: Creating the BAM file and the Transcript from reads and index files
Step 2 is similar to Step 1.
From folder step1, to run step 2:
$ cd ..
$ cd step2
$ snakemake
After Step 2:
rnaseq-snakemake
|_________data
| |_________ggal_gut_1.fq
| |_________ggal_gut_2.fq
| |_________ggal.gtf
| |_________reference.fa
|_________step1
| |_________indexing.smk
| |_________Snakefile
| |_________index.done
| |_________reference
| |_________index.1.ht2
| | (2-7)
| |_________index.8.ht2
|_________step2
| |_________therest.smk
| |_________Snakefile
| |_________[ggal_gut.cutadapt.sam]
| |_________[e2t.ctab]
| |_________[i_data.ctab]
| |_________[i2t.ctab]
| |_________[t_data.ctab]
| |_________[e_data.ctab]
| |_________[ggal_gut.tsv]
| |_________[ggal_gut_ref.gtf]
| |_________[ggal_gut_transcript.gtf]
| |_________[ggal_gut.cutadapt.bam]
| |_________[ggal_gut.cutadapt.bam.bai]
|_________environment.yml
The script will call hisat2 , samtools, and stringtie to do the work. The same principle as step 1 applies to this step, the Snakefile will contains the final outputs, and call to the helper script therest.smk. ggal_gut.cutadapt.sam file will contains the sequence alignment data produced by mapping reads to the reference genome. ggal_gut.cutadapt.bam file will contain the compressed binary data from Sam. More description on ctab files, gtf outputs, and tsv of stringtie can be found here. The ggal_gut_transcript.gtf contains details of the transcripts that StringTie assembles from RNA-Seq data, while ggal_gut.tsv contains gene abundances.
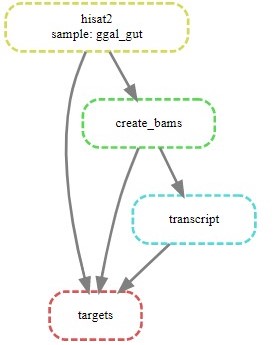
Creating a visualization for your workflow
In the step2 folder:
$ snakemake --dag | dot -Tsvg > visual.svg
A file named visual.svg will be created in the same folder; it can be downloaded and open with any web browser. It should look like this:
About environment.yml
channels:
- conda-forge
- bioconda
- main
- r
dependencies:
#snakemake and python will be included
- snakemake-minimal =5.10.0
- python =3.7.6
#all other bioinformatics tools
- samtools =1.9
- bowtie2 =2.3.5.1
- hisat2 =2.2.0
- stringtie =2.1.2
- gffread =0.11.7
#visualization tool
- graphviz =2.42.3e
To see the result of this workflow, you can check it here.