Running Nextflow RNA-seq
This Nextflow RNA-seq workflow maps read-pairs to a reference genome and produces a transcript.
By using software containers, Nextflow enables scalable and reproducible scientific workflows. Pipelines can be written in any common scripting language.
Requirements:
Docker
Java (required to install Nextflow)
Nextflow
Graphviz
To install Docker, Graphviz and Nextflow, see our VM Workflow Tools Installation Cheatsheet for instructions.
Note
The requirements above are crucial to running this workflow. Please make sure you have them installed properly prior to running this workflow.
Download this tutorial:
$ sudo add-apt-repository universe
$ sudo apt update
$ sudo apt install subversion
#cloning this tutorial
$ svn checkout https://github.com/isb-cgc/RunningWorkflows-on-the-GoogleCloud/trunk/Nextflow-RNAseq
Running Nextflow
You should have a Nextflow-RNAseq directory :
Nextflow-RNAseq
├── data
│ ├── sample_1.fq
│ ├── sample_2.fq
│ ├── sample.fa
│ └── sample.gtf
├── main.nf
└── nextflow.config
The file main.nf contains all the code to execute the workflow, and nextflow.config provides the name of the Docker image that contains all of the tools for this run.
Let’s take a look at the contents of the file main.nf. The first block contains all the parameters (denoted by “params”) that are required to input into the workflow.
#!/usr/bin/env nextflow
params.outdir = "$baseDir/results"
params.indexDir = "$baseDir/reference"
params.refGenome = "$baseDir/data/sample.fa"
params.forRead = "$baseDir/data/sample_1.fq"
params.revRead = "$baseDir/data/sample_2.fq"
params.gtfFile = "$baseDir/data/sample.gtf"
The process block contains 3 basic parts: input, output, and command (contained in the pairs of quotations “”” some commands “””). The variable refGenome with type path is created to hold the value of params.refGenome.
In the command (“””) block, the hisat2 build command will be executed with the previously created variable refGenome denoted as ${refGenome}. The name of the final output files from hisat2 build in this case will follow the pattern index.#.ht2.
In the output section, in order to catch the outputs of hisat2, the wildcard ‘index*’ was used, and those outputs will become a list assigned to the channel index_ch* variable. The index_ch* channel can be used by downstream processes. The other 3 processes have similar syntax.
process buildIndex {
publishDir params.indexDir, mode: 'copy'
echo true
input:
path refGenome from params.refGenome
output:
path 'index*' into index_ch
"""
echo "Building Indices"
hisat2-build ${refGenome} index
"""
}
To run:
#Assume the executable file "nextflow" is installed in the same directory with the folder you downloaded "Nextflow-RNAseq".
$ ./nextflow run Nextflow-RNAseq
After the workflow finishes running, the folder should look like this:
Nextflow-RNAseq
├── data
│ ├── sample_1.fq
│ ├── sample_2.fq
│ ├── sample.fa
│ └── sample.gtf
├── main.nf
├── nextflow.config
├── [reference]
│ ├── [index.1.ht2]
│ ├── [index.2.ht2]
│ ├── [index.3.ht2]
│ ├── [index.4.ht2]
│ ├── [index.5.ht2]
│ ├── [index.6.ht2]
│ ├── [index.7.ht2]
│ └── [index.8.ht2]
└── [results]
├── [final_ref.gtf]
├── [final_transcript.gtf]
├── [sample.bam]
├── [sample.sam]
└── [sample.tsv]
The script will call hisat2 , samtools, and stringtie to do the work. sample.sam file will contain the sequence alignment data produced by mapping reads to the reference genome, sample.bam file will contain the compressed binary alignment data. Additional information about the gtf and tsv outputs of StringTie can be found here. The final_transcript.gtf contains details of the transcripts that StringTie assembles from RNA-Seq data, while final.tsv contains gene abundances.
Running Nextflow with visualization
Use the following command:
#Assume the executable file "nextflow" is installed in the same directory with the folder you downloaded "Nextflow-RNAseq".
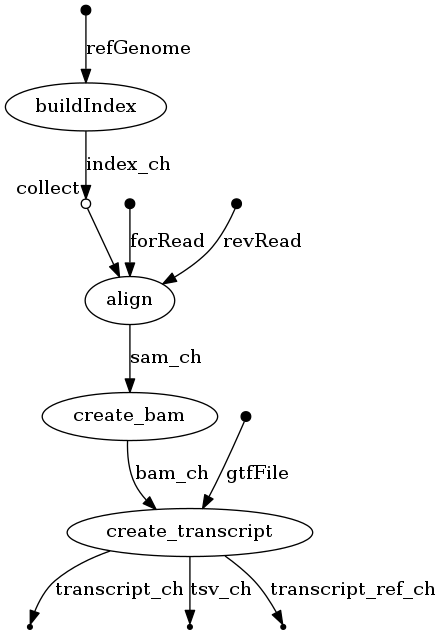
$ ./nextflow run Nextflow-RNAseq -with-dag flowchart.png
An image file with the name flowchart.png will be available to download. It should look like this:
To see the result of this workflow, you can check it here.