Running Nextflow pipeline on public BAM file from ISB-CGC
This Nextflow workflow gathers GC content from a BAM file (or a list of BAM files) to a text file. By using software containers, Nextflow enables scalable and reproducible scientific workflows. Pipelines can be written in the most common scripting languages.
Requirements:
Docker
Java (required to install Nextflow)
Gcsfuse
Nextflow
A public bam file from ISB-CGC at the address: gs://gdc-ccle-open/692a845c-7957-41f2-b679-5434c69ba25b/G27328.Calu-6.1.bam
To install Docker and Nextflow, see our VM Workflow Tools Installation Cheatsheet for instructions. To set up gcsfuse in order to get access to the BAM file, please visit Running Workflow with GCSFUSE.
Note
The requirements above are crucial to running this workflow. Please make sure you have them installed properly prior to running this workflow.
Download this tutorial:
$ sudo add-apt-repository universe
$ sudo apt update
$ sudo apt install subversion
#cloning this tutorial
$ svn checkout https://github.com/isb-cgc/RunningWorkflows-on-the-GoogleCloud/trunk/Nextflow-GCgather
Running Nextflow
You should have a Nextflow-GCgather directory with one file called Nextflow-GCgather.nf inside. We are going to change the address in this file to the one you created in the Running Workflow with GCSFUSE tutorial.
#go into the folder
$ cd Nextflow-GCgather
$ nano Nextflow-GCgather.nf
At the top of the file you will see this:
#!/usr/bin/env nextflow
myBamSample = Channel.fromPath('/home/thinh_vo/sample/*.bam')
Replace “/home/thinh_vo/sample/*.bam” with your new address from the gcsfuse tutorial for example: “/home/thinh_vo/testGcsfuse/*.bam”. Now the script is ready to run with Nextflow.
#Go to where the Nextflow executable file was installed in this example. It will be outside the Nextflow-GCgather directory.
#First, we get out of Nextflow-GCgather directory.
$ cd ..
#execute nextflow with docker image:
$ ./nextflow run Nextflow-GCgather/Nextflow-GCgather.nf -with-docker gcr.io/genomics-tools/samtools
Note
This BAM file is quite large; it may take about 15 mins ~ 20 mins to run.
Once Nextflow is finished, the result will be on the screen, or you can find it at Nextflow-GCgather/Sam_results/final_gc_stats_out.txt.
Running Nextflow with visualization
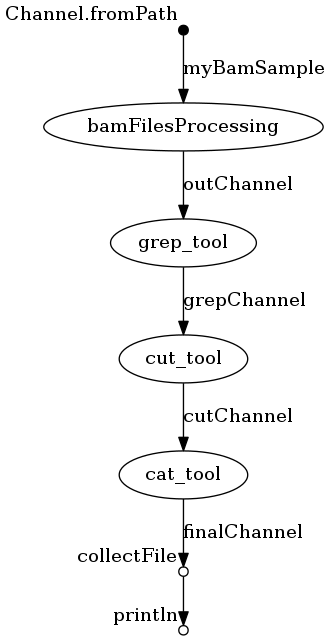
You can use this command instead to run Nextflow; it will out put a visualization file named “flowchart.png”.
$ ./nextflow run Nextflow-GCgather/Nextflow-GCgather.nf -with-dag flowchart.png
It should look like this:
To see the result of this workflow, you can check it here.